Patient With Hypertension Undergoes Uncommon Surgery
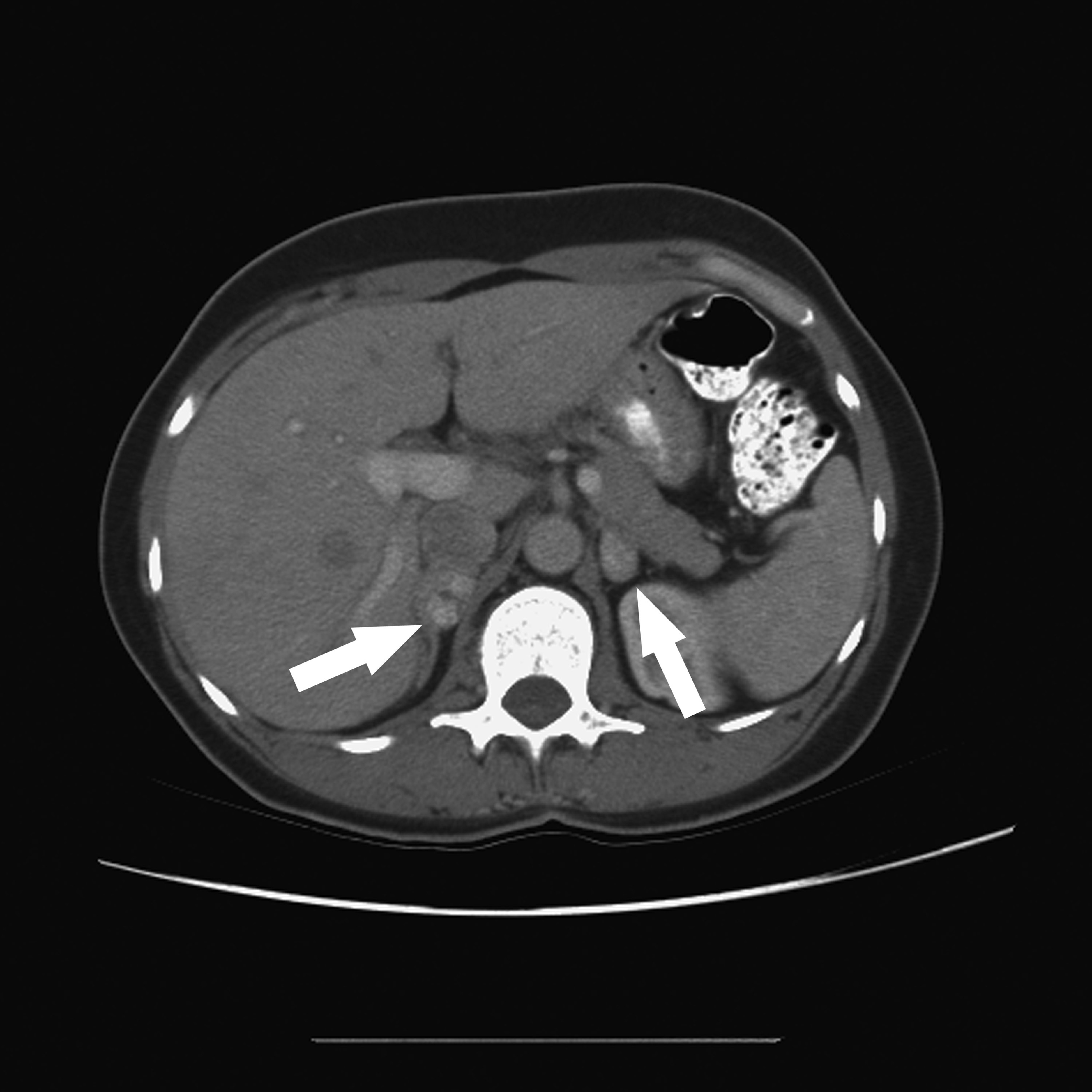
Following the discovery of persistent, severe hypertension, a 22-year-old man in college was referred to Duke Health’s nationally recognized endocrine neoplasia program with a likely diagnosis of bilateral pheochromocytomas.
Julie A. Sosa, MD, an endocrine surgeon, surgical oncologist, and director of the Duke Health endocrine neoplasia disease program, and her team developed a comprehensive treatment plan for the patient. He underwent complex, bilateral, retroperitoneal laparoscopic adrenalectomy.
The procedure was successful, and the patient recovered quickly and began his postgraduate professional studies.
As part of a comprehensive preoperative approach, his health care team at Duke initiated genetic screening. The results of these tests yielded an important discovery.
QUESTION: What did the endocrinologists and surgeons learn from the genetic test results?
View case conclusion
MAX mutations are responsible for 1.1% of pheochromocytomas and paragangliomas in persons without evidence of other mutations.
Because the MAX mutation suggests hereditary disease, the patient’s 57-year-old father was also tested and was found to have the same genetic mutation. A review of his medical records also identified a pattern of mild but persistent hypertension, a key clue to the presence of pheochromocytoma.
Similar to his son, the father underwent retroperitoneal laparoscopic adrenalectomy to remove his left adrenal gland. This relatively new, minimally invasive procedure to remove 1 or both adrenal glands through small incisions in the back was used to minimize potential deleterious effects on the patient's other organs, such as bowel injury and ileus, and to shorten the time needed for the operation.
Retroperitoneal laparoscopic adrenalectomy is also associated with faster patient recovery, shorter hospital stays, and reduced levels of pain.
The members of the Hereditary Cancer Center at the Duke Cancer Institute were consulted when developing the transdisciplinary care directed by the endocrine neoplasia team. Sosa says that the health care collaboration among her multidisciplinary team at Duke, like what was seen with this patient, is key when encountering a relatively uncommon, complex case.
“This is the distinction of our care at Duke Health,” Sosa adds. “We thought early on about the potential for a rare inherited syndrome, performed a surgical procedure that enhanced patient outcomes, and coordinated genetic screening that allowed us to intervene early with surgery.”
Retroperitoneal laparoscopic adrenalectomies for the son and his father were performed within 2 years of each other. Sosa notes that both family members responded well to the procedures and were quick to recover.
“The discovery of the MAX mutation helped us resolve a potentially life-threatening condition,” says Sosa.
The endocrine neoplasia group includes 40 specialists, including clinicians and researchers, who manage the full spectrum of endocrine neoplasia disorders, including benign and malignant conditions of the pancreas and thyroid, parathyroid, and adrenal glands. The group performs 50 or more retroperitoneal laparoscopic adrenalectomies every year. Sosa’s team also conducts clinical trials.